PAPEL DEL OXIDO
NITRICO EN LOS FENOMENOS INFLAMATORIOS DE LAS MUCOSAS.
Dr. Fernando Soteras Abril
Facultad de Medicina. Universidad dee Zaragoza.
INTRODUCCION.
El monóxido
de nitrógeno, más comúnmente llamado óxido nítrico o simplemente NO es
considerado funcionalmente como un radical libre, aunque su estructura
electrónica no corresponde a la de estos elementos. Este hecho es debido a sus
propiedades y sobretodo a los derivados que surgen en su metabolismo todos
ellos con estructura y función de radical libre.
Derivados
del NO
|
|
Dióxido de nitrógeno (NO2) Trióxido de Dinitrógeno (N2O3) Tetraóxido de Dinitrógeno
(N2O4) Peroxinitritos (ONOO-) |
Tabla 1. Derivados del NO con acciones de radical
libre.
El NO fue
inicialmente detectado como un mediador endógeno liberado desde las células endoteliales con
efecto en la relajación vascular (Palmer et al., 1987) y como inhibidor de la agregación
plaquetaria y adhesión de los neutrófilos (Kubes et al., 1991).
El NO es
una pequeña molécula (30 Da) difusible en líquidos y tejidos corporales, con
una vida media muy corta (de pocos segundos a algún minuto), sintetizada desde
el nitrógeno terminal de la L-Arginina. Su síntesis es catalizada por la óxido
nítrico sintasa (NOS) usando los siguientes cofactores: Flavin mononucleotido,
flavin adenin dinucleotido, tetrahidrobiopterina y protoporfirina IX. Se han
caracterizado tres diferentes isoformas de esta enzima de acuerdo al origen
celular y características bioquímicas. Dos enzimas constitutivas (cNOS) una de
origen neuronal (nNOS) o tipo 1 y otra endotelial (eNOS) o tipo 3 y una tercera
inducible (iNOS) o tipo 2. Las dos primeras son calcio/calmodulina dependientes
y la última es independiente de la presencia de este elemento. Estas isoenzimas
son codificadas por tres genes diferentes lo que conlleva características
diferenciadas (Forstermann and Kleinert, 1995).
La inhibición de la NOS puede realizarse mediante
sustancias que eliminen o contrarresten a la calmodulina, flavoproteínas,
grupos hemo y tetrahidrobiopterina; sin embargo, los más usados son los
análogos a la L-Arginina como son NG-metil-L-arginina
(L-NMMA), NG-nitro-L-arginina
(L-NNA) y NG-nitro-L-arginina-metilester
(L-NAME) que inhiben a las tres isoenzimas con diferente intensidad.
Actualmente se busca una inhibición selectiva y se tiene para la nNOS con el
7-nitroindazol y para la iNOS con aminoguanidina, L-N6-(1-iminoetil)lisina y
derivados de la isotiourea (Hobbs et al., 1999).
REGULACION
DE LAS ISOFORMAS DE LA NOS.
La producción de NO por las células endoteliales se
produce cuando se incrementa el calcio intracelular. Este se une a la proteína
calmodulina formando un complejo que se une a su vez a la enzima eNOS. Esta
síntesis constitutiva genera pequeñas cantidades de NO el cual participa en
procesos fisiológicos celulares junto al GMPc (Moncada et al., 1991).
Mecanismos como el estrés y la acción de los estrógenos se han descrito como
reguladores de la acción de la eNOS de forma independiente del calcio (Dimmeler
et al., 1999).
En las neuronas sucede algo similar, la llegada de un
potencial de acción a una neurona activa los canales de calcio en el neurolema
permitiendo la entrada de este ión dentro de la célula, su unión a la
calmodulina y posteriormente a la nNOS. Aunque la enzima es considerada como
constitutiva, en situaciones de lesión nerviosa puede modularse su actividad
(Steel et al., 1994).
En contraste a las dos isoformas constitutivas, la iNOS
es calcio-independiente. En situación fisiológica la actividad de esta enzima
es muy baja o ausente; sin embargo, cuando es inducida genera grandes
cantidades de NO de forma mantenida interviniendo en múltiples acciones con
infección o inflamación (Moncada et al., 1991). La actividad iNOS se incrementa
por el lipopolisacárido bacteriano (LPS), IL-1, IFN-g, TNF-a, factor de crecimiento
plaquetario, factor de crecimiento de fibroblasto, agentes estimulantes de la
protein-kinasa-C y del AMPc. Existen otros factores que la inhiben como son la
IL-4 y la IL-8 entre otros.
Existe una evidencia reciente que sugiere una estrecha
interacción entre eNOS e iNOS lo que indica un complejo mecanismo de producción
y regulación de NO (Colasanti and Suzuki, 2000).
ACCIONES FISIOLOGICAS DEL NO.
La mayor parte de las células del organismo son capaces
de producir NO en un momento determinado ya sea de forma constitutiva o a
consecuencia de una agresión. Esta multiplicidad de orígenes hace que sus
funciones fisiológicas sean a su vez muy variadas. Por sistemas a nivel
vascular participa en la regulación de la circulación sanguínea, en el control
de la presión arterial, inhibe la agregación plaquetaria así como la adhesión
de los neutrófilos. En el sistema nervioso central participa en la coordinación
entre la actividad neuronal y el débito sanguíneo así como en la modulación del
dolor. En el sistema nervioso periférico es un neurotransmisor sináptico no
adrenérgico no colinérgico. En las mucosas incrementa la vascularización y
producción de moco y por lo tanto en la integridad de la misma. En el pulmón su
acción es variada y en algunos casos perjudicial; así el NO de origen
endotelial regula la circulación pulmonar, el neuronal tiene acciones broncodilatadoras
y el producido por la iNOS puede provocar asma, bronquiectasia y fibrosis
alveolar (Calatayud et al., 2001; Kubes, 2000).
INTEGRIDAD
DE LA MUCOSA Y OXIDO NITRICO.
La mucosa está continuamente expuesta a agresores y su
capacidad de resistencia está basada en un proceso dinámico que implica a la
secreción epitelial, la función de barrera, la microcirculación y al sistema
inmune. Diversos mediadores de carácter endógeno participan en la integridad de
la mucosa; no obstante, el papel del NO de forma aislada o regulando a otros
mecanismos no esta aclarado completamente.
Inicialmente se pensaba que la disminución del NO
procedente de la cNOS o el incremento de NO producido por la iNOS provocaba
lesión de la mucosa.
|
Figura 1. Esquema
inicial del origen de la lesión por el NO.
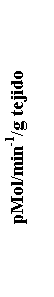
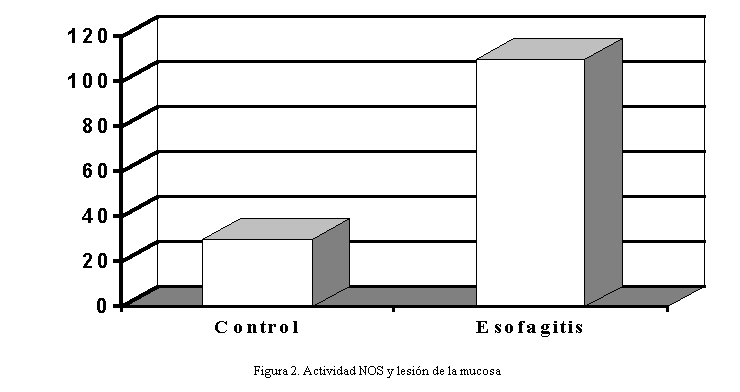 En estudios
realizados por nuestro grupo se observó que una agresión externa y moderada de
la mucosa (esófago) produce un incremento en la actividad NOS que no siempre se
corresponde con un aumento equivalente de la actividad iNOS (Figura 2);
asimismo, en este mismo tipo de agresión la administración de un inhibidor de
la actividad NOS aumenta el grado de lesión (Soteras et al., 2000).
En estudios
realizados por nuestro grupo se observó que una agresión externa y moderada de
la mucosa (esófago) produce un incremento en la actividad NOS que no siempre se
corresponde con un aumento equivalente de la actividad iNOS (Figura 2);
asimismo, en este mismo tipo de agresión la administración de un inhibidor de
la actividad NOS aumenta el grado de lesión (Soteras et al., 2000).
Por otro lado, en
agresiones intensas de la mucosa (esófago) se apreció un incremento en la
actividad NOS con elevación de la actividad iNOS que descendió progresivamente
conforme se incrementaba el daño, en esta situación se detectaron niveles de
peroxinitritos. La administración de agentes que modifican la actividad NOS no
produjo cambios sustanciales en el daño mucosa mientras que, la administración
de superóxido dismutasa (SOD) previno la lesión (Lanas et al., 2001).
Estos hechos indican
que el papel protector del NO en la mucosa constituye un mecanismo más complejo
que el simple esquema representado inicialmente en la Figura 1.
En términos generales podríamos señalar que a bajas concentraciones el
NO es pro-inflamatorio favoreciendo la vasodilatación y el reclutamiento de
neutrófilos mientras que a concentraciones elevadas disminuye la capacidad de
adhesión celular e induce apoptosis de las células inflamatorias (Ross et al.,
2001).
Baja
Cocentración
|
Alta
Concentración
|
|
Pro-inflamatorio Vasodilatador
Reclutamiento de
neutrófilos |
Disminución moléculas
adhesión. Suprime la activación
celular Apoptosis células
inflamatorias |
Tabla 2. Efectos generales del NO
en el sistema inmune.
El reclutamiento de
los leucocitos hacia las áreas inflamatorias continuando con su adhesión a las
células del endotelio vascular seguido de la emigración hacia el compartimento
intersticial constituye un hecho histológico cardinal en las enfermedades
inflamatorias de la mucosa (Elliott and Wallace, 1998).
La implicación del
NO fue observada inicialmente cuando se produjo una elevada adherencia de
leucocitos al endotelio tras la inhibición de la NOS un proceso que depende de
las a2-integrinas (Kubes
et al., 1991) y P-selectina (Davenpeck et al., 1994). Se ha observado en
ratones con déficits en la síntesis de iNOS una mayor capacidad de adhesión de
leucocitos a las células del endotelio vascular lo que sugiere que el NO en los
procesos inflamatorios efectúa un control homeostático que frena el
reclutamiento leucocitario hacia los tejidos inflamados (Hickey et al., 1997).
Los factores que desencadenan la adherencia de los neutrófilos también
provocan la activación de estas células y la liberación de radicales libres
capaces de dañar el endotelio vascular; además, esta adherencia puede
dificultar el flujo sanguíneo debido a obstrucciones de los capilares con la
consiguiente isquemia y disminución de la capacidad defensiva de la mucosa.
Para prevenir en una primera etapa esta lesión endotelial el NO podría
disminuir este reclutamiento y adherencia leucocitaria y junto a otras
substancias vasodilatadoras, prostaciclinas, favorecer el aporte sanguíneo a
dicha mucosa. Por otra parte, el NO es capaz de inhibir la actividad de los
mastocitos y de la producción de substancias pro-inflamatorias por parte de
ellos. Además, inhibe la agregación plaquetaria lo que favorece la acción
anti-inflamatoria de este elemento.
En el sistema
inmunológico, el NO participa de diversas formas con acciones tan diversas como
agente tóxico frente a organismos infecciosos (Hibbs et al., 1987), inductor o
supresor de la apoptosis (Kröncke et al., 2001) o inmunorregulador (Wei et al.,
1995).
Debido a las características del sistema inmune cuya
capacidad de reacción se prolonga de días a semanas, la producción de NO debe
de realizarse de forma intensa y continuada y es la isoforma iNOS la que entra
en actividad en esta situación (Coleman, 2001).
Muchos tipos de células inflamatorias e inmunes expresan
actividad iNOS incluyendo monocitos/macrófagos (Stuehr et al., 1985), células
presentadoras de antígenos (van der Veen, 2001) y células “asesinas” o “natural
killer” (células NK) (Cifone et al., 2001). Sin embargo en otros tipos
celulares como neutrófilos, mastocitos o linfocitos T se cuestiona su presencia
(van der Veen et al., 2001; Forsythe et al., 2001; Armstrong, 2001).
A nivel experimental
el NO de origen macrofágico presentó en cultivo una inhibición de la
proliferación de los linfocitos T (van der Veen et al., 2001). Estudios
posteriores en ratones has mostrado que el NO inhibe selectivamente a los
linfocitos Th1 y de una manera autocrina promueve la respuesta de los Th2
conduciendo de esta manera a la producción de IgE y de las enfermedades en la
que esta implicada esta inmunoglobulina (Barnes and Liew, 1995). No obstante
estos estudios están cuestionados por otros autores y en el ser humano se han
descrito incluso inhibiciones de ambas estirpes celulares por el NO (Bauer et
al., 1997).
El NO es un mediador de la actividad y función de las
células NK, inhibe la activación de los mastocitos y puede elevar o inhibir la
activación de los neutrófilos en dependencia de su concentración.
En conclusión
empezamos a conocer algunos mecanismos en los que interviene esta molécula no
obstante, queda aun mucho por descubrir y serán precisas más experiencias para
llegar a entender completamente la participación del NO en la inflamación y su
posterior utilización como medio terapeútico.
BIBLIOGRAFIA.
Armstrong
R. 2001. The physiological role and pharmacological potential of nitric oxide
in neutrophil activation. Int Immunopharmacol 1:1501-1512.
Barnes
PJ, Liew FY. 1995. Nitric oxide and asthamatic inflammation. Immunol Today
16:128-130.
Bauer H, Jung T,
Tsikas T, Stichtenoth DO, Frolich JC, Neumann C. 1997. Nitric oxide inhibits
the secretion of T-helper 1 and T-helper 2 associated cytokines in activated T
cells. Immunology 90:205-211.
Calatayud
S, Barrachina MD, Esplugues JV. 2001. Nitric oxide: relation to integrity,
injury and healing of the gastric mucosa. Microsc Res Tech 53:325-335.
Cifone
MG, Ulisse S, Santoni A. 2001. Natural killer cells and nitric oxide. Int
Immunopharmacol 1:1513-1524.
Colasanti
M, Suzuki H. 2000. The dual personality of NO. Trends Pharmacol Sci 21:249-252.
Coleman
JW. 2001. Nitric oxide in immunity and inflammation. Int Immunopharmacol
1:1397-1406.
Davenpeck
KL, Gauthier TW, Lefer AM. 1994. Inhibition of endothelial-derived nitric oxide
promotes P-selectin expression and actions in the rat microcirculation.
Gastroenterology 107:1050-1058.
Dimmeler
S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. 1999. Actication of
nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation.
Nature 399:601-605.
Elliott
SN, Wallace JL. 1998. Neutrophil-mediated gastrointestinal injury. Can J
Gastroenterology 12:559-568.
Forstermann
U, Kleinert H. 1995. Nitric oxide synthase: expression and expressional control
of the three isoforms. Naunyn Schmiedebergs Arch Pharmacol 352:351-364.
Forsythe
P, Gilchrist M, Kulka M, Befus AD. 2001. Mast cells and nitric oxide: control
of production, mechanism of response. Int Immunopharmacol 1:1524-1541.
Hibbs
JB, Taintor RR, Vavrin Z. 1987. Macrophage cytotoxicity role for L-arginine
deiminase and imino nitrogen oxidation to nitrite. Science 235:473-476.
Hickey
MJ, Sharkey KA, Sihota EG, Reinhardt PH, Macmicking JD, Nathan C, Kubes P.
1997. Inducible nitric oxide synthase-deficient mice have enhanced
leukocyte-endothelium interactions in endotoxemia. FASEB J 11:955-964.
Hobbs
AJ, Higgs A, Moncada S. 1999. Inhibition of nitric oxide synthase as a
potential therapeutic target. Annu Rev Pharmacol Toxicol 39:191-220.
Kröncke
KD, Fehsel K, Susehek C, Kolb-Bachofen V. Inducible nitric oxide
synthase-derived nitric oxide in gene regulation, cell death and cell survival.
Int Immunopharmacol 1:1407-1420.
Kubes P. 2000.
Inducible nitric oxide synthase: a little bit of good in all of us. Gut 47:6-9.
Kubes
P, Suzuki M, Granger DN. 1991. Nitric oxide: an endogenous modulator of
leukocyte adhesion. Proc Natl Acad Sci USA 88:4651-4655.
Lanas A, Soteras F, Jimenez
P, Fiteni I, Piazuelo, E, Royo Y, Ortego J, Iñarrea P, Esteva F. 2001. Superoxide anion and nitric oxide in high-grade esophagitis induced by
acid and pepsin in rabbits. Dig Dis Sci 46:2733-2743.
Moncada
S, Palmer RM, Higgs EA. 1991. Nitric oxide: physiology, pathophysiology and
pharmacology. Pharmacol Rev 43:109-142.
Palmer RM, Ferrige AG, Moncada
S. 1987. Nitric oxide release accounts for the
biological activity of endothelium-derived relaxing factor. Nature 327:524-526.
Ross
R, Reske-Kuntz AB. 2001. The role of nitric oxide in contact hypersensitivity. Int Immunopharmacol
1:1469-1478.
Soteras F, Lanas A, Fiteni
I, Royo Y, Jimenez P, Iñarrea P, Ortego J, Esteva F. 2000. Nitric oxide and superoxide anion in low grade esophagitis induced by
acid and pepsin in rabbits. Dig Dis Sci 45:1802-1809.
Steel
JH, Terenghi G, Chung JM, Na HS, Carlton SM, Polak JM. 1994. Increased nitric
oxide synthase immunoreactivity in rat dorsal root ganglia in a neuropathic
pain model. Neurosci Lett 169:81-84.
Stuehr
DJ, Marlette MA. ¡985. Mammalian nitrate biosynthesis: mouse macrophages
produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide.
Proc
Natl Acad Sci USA 82:7738-7742.
van
der Veen RC. 2001.Nitric oxide and T helper cell immunity. Int Immunopharmacol
1:1491-1500.
Wei
X, Charles IG, Smith A, Ure J, Feng G, Huang F. 1995. Altered immune responses
in mice lacking inducible nitric oxide synthase. Nature 375:408-411.